Membrane Lipids and Steroids
Cholesterol is transported in the blood by LDLs and taken up into cells by the LDL receptor on the cell surface. The LDL receptor is absent in people with familial hypercholesterolemia.
Phospholipids and Triacylglycerols
The first step in the synthesis of both phospholipids and triacylglycerols is the synthesis of phosphatidate. Dihydroxyacetone phosphate can be reduced to glycerol-3-phosphate, which then undergoes the addition of two fatty acids to form phosphatidate. The addition of fatty acids is catalyzed by glycerol phosphate acyltransferase. The pathways then diverge at phosphatidate.
In the synthesis of triacylglycerols, phosphatidate is hydrolyzed by a specific phosphatase to give diacyglycerol. This intermediate is acylated to triacylglycerol by diglyceride acyltransferase. The liver is the primary site of triacylglycerol synthesis, from the liver, triacyglycerols are sent to muscle for conversion to energy or to adipose tissue for storage.
Phospholipid synthesis continues in the endoplasmic reticulum. Phospholipid synthesis requires the combination of a diacylglycerol with an alcohol, and the diacylglycerol or the alcohol must be activated first.
Cholesterol
Cholesterol modulates fluidity in the cell membrane and is the precursor of steroid hormones such as progesterone, testosterone, estradiol, and cortisol. All 27 carbon atoms of cholesterol are derived from acetyl CoA in a three step process:
(1) the synthesis of isopentenyl pyrophosphate (cytoplasm)
(2) the condensation of six molecules of isopentenyl pyrophosphate to form squalene (endoplasmic reticulum)
(3) cyclization of squalene (endoplasmic reticulum)
The first stage of cholesterol synthesis is the formation of isopentenyl pyrophosphate from acetyl CoA. This stage begins with the formation of HMG-CoA (note: this molecule was also formed in ketone body formation) from acetyl CoA and acetoacetyl CoA. HMG-CoA is then reduced to mevalonic acid by HMG-CoA reductase. The synthesis of mevalonic acid is the committed step in cholesterol formation. HMG-CoA reductase is an integral membrane protein in the endoplasmic reticulum. Mevalonic acid in the converted to 3-isopentenyl pyrophosphate in three consecutive reactions, each requiring a molecule of ATP. Thus, 3 ATPs are needed to convert mevalonic acid to 3-isopentenyl pyrophosphate. One molecule of carbon dioxide is also produce in the last of these reactions.
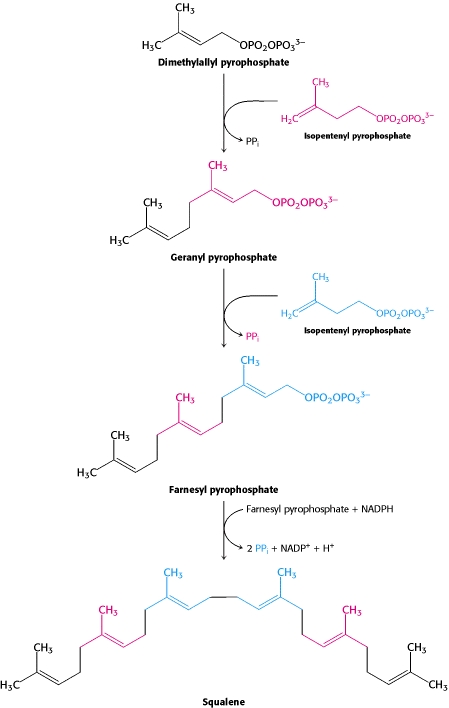
One molecule of dimethyallyl pyrophosphate and two molecules of isopentenyl pyrophosphate condense to form farnesyl pyrophosphate. The tail-to-tail coupling of two molecules of farnesyl pyrophosphate yields squalene.
(c) 2007 W.H. Freeman and Company
Squalene is activated by conversion into squalene epoxide, this reaction uses oxygen and NADPH. Squalene is then converted to lanosterol which is converted to cholesterol by the removal of three methyl groups, and the reduction of double bond by NADPH.
Regulation of Cholesterol Synthesis
The liver is the major site of cholesterol synthesis in mammals, and the rate of cholesterol synthesis is highly responsive to the cellular level of cholesterol. This feedback regulation is mediated primarily by changes in the amount and activity of HMG-CoA reductase. HMG-CoA reductase is controlled in multiple ways:
(1) The rate of synthesis of reductase mRNA is controlled by the sterol regulator element binding protein (SREBP), which binds to the sterol regulatory element (SRE) when cholesterol levels are low to initiate transcription of HMG-CoA reductase.
(2) The rate of translation of HMG-CoA reductase is inhibited by nonsterol metabolites derived from mevalonate and cholesterol.
(3) HMG-CoA reductase has a cytoplasmic region which carries out catalysis, and a membrane region which senses signals that lead to the degradation of HMG-CoA reductase. The reductase can also be degraded by ubiquination and targeting to the 26S proteasome. Also the enzyme may be made more susceptible to proteolysis.
(4) Phosphorylation decreases the activity of the reductase. HMG-CoA reductase, like acetyl CoA carboxylase (which catalyzes the irreversible step in fatty acid synthesis) is switched off by AMP-dependent protein kinase. Thus cholesterol synthesis ceases when ATP is low.
Transport of Cholesterol
Cholesterols and triacylglycerols are transported in the form of lipoprotein particles in body fluids. The protein components of these molecules are called apoproteins. Apoproteins have two roles: they solubilize hydrophobic lipids and contain cell targeting signals. Apolipoproteins are synthesized and secreted by the liver and the intestine.
Triacylglycerols, cholesterol, and other lipids are carried away from the intestine in the form of chylomicrons. Apolipoprotein B-48, a large protein, forms an amphipathic spherical shell around the fat globule. Lipoprotein lipase is found lining the blood vessels in muscle and other tissues that use fatty acids as energy. Lipoprotein lipases release triacylglycerols from the chylomicrons by hydrolysis. The liver then takes up the cholesterol-rich residues, known as chylomicron remnants.
The liver is the major site of triacylglycerol and cholesterol synthesis. Triacylglycerols and cholesterol in excess of the liver's own needs are exported into the blood in the form of VLDLs. VLDLs are stabilized by apo B-100 and apo E. Triacylglycerols in VLDLs are hydrolyzed by lipases on capillary walls. The resulting remnants, rich in cholesteryl esters, are IDLs.
IDLs can either be taken up by the liver for processing or they can be converted into LDLs by the removal of more triacylglycerol.
LDL is the major carrier of cholesterol in blood. LDL contains a single copy of apo B-100, which is recognized by target cells. The role of LDL is to transport cholesterol to peripheral tissues and regulate de novo cholesterol synthesis at these sites.
In contrast, HDL picks up cholesterol released into plasma by dying cells and from membranes undergoing turnover. An acyltransferase esterifies these cholesterols, which are then returned to the liver by HDL.
LDLs
We have already seen that cholesterol synthesis in the liver is controlled by the amount of dietary cholesterol, which reduces the amount and activity of HMG-CoA reductase.
In general, cells outside the liver and intestine obtain cholesterol from the plasma, specifically, from LDL. The process of LDL uptake is called receptor mediated endocytosis.
Steps of receptor mediated endocytosis include:
(1) apo B-100 on the surface of LDL particle binds to a receptor on the plasma membrane of nonhepatic cells.
(2) the receptor LDL complex is internalized by endocytosis forming vesicles.
(3) these vesicles containing LDL, subsequently fuse with lysosomes. The protein component of LDL is hydrolzed by the degradative enzymes in the lysosome to free amino acids. The cholesteryl esters are hydrolyzed by lysosomal acid lipase.
(4) The released unesterified cholesterol molecules can then be used for membrane biosynthesis. Alternatively, the cholesterol can be reesterified for storage within the cell. High concentrations of unesterified cholesterol disrupts the integrity of the cell membrane.
Studies have also shown that when cholesterol is abundant inside the cell, new LD receptors are not synthesized, and so the uptake of additional cholesterol from the plasma LDL is blocked.
In familial hypercholesterolemia, cholesterol is deposited in various tissues because of the high concentration of LDL cholesterol in the plasma. Excess blood LDL can also be oxidized to form oxLDL, which is taken up the the immune system (macrophages), which become engorged to form foam cells. Foam cells become trapped in the walls of vessels and contribute to the formation of plaques, which cause narrowing of the arteries. The defect in most cases with familial hypercholesterolemia is the absense of LDL receptors. Thus, the entry of LDL into the liver and other cells in impaired, leading to an increased level of LDL in plasma. Furthermore, less IDL enters the liver, because IDL entry is too controlled by the LDL receptor. Therefore, IDL stays in the plasma longer and more of it is converted to LDL. .
Management of Cholesterol
The only treatment for homozygous familial hypercholesterolemia is liver transplant. In heterozygotes and others with high cholesterol, the goal is to reduce the amount of cholesterol in the blood by stimulating the gene to produce more than the customary number of LDL receptors. Since the production of LDL receptors is controlled by the cells need for cholesterol, the strategy is to deprive the cell fo ready sources of cholesterol. When cholesterol is then required, the amount of mRNA for the LDL receptor will be increased. This can be done is two ways: (1) reabsorption of bile salts from the intestine is inhibited. Because bile salts are cholesterol derivatives that promote the aborption of cholesterol and fats more of them in the blood will stimulate more absorption of cholesterol. (2) de novo synthesis of cholesterol is blocked.
Positively charged polymers are administered orally to inhibit absorption of bile salts.
Cholesterol synthesis can be effectively blocked by statins. Statins are potent competitive inhibitors of HMG-CoA reductase.